Фенилкетонурия
Фенилкетонурия: симптомы и лечение

Фенилкетонурия (фенилпировиноградная олигофрения, болезнь Феллинга) – это наследственное заболевание, связанное с нарушением обмена аминокислоты фенилаланина. В результате накопления токсических продуктов из-за неправильного метаболизма развивается отставание в умственном и физическом развитии.
Причем появившиеся нарушения в состоянии здоровья необратимы, но своевременная диагностика заболевания может предотвратить все патологические изменения. Лечение заключается в исключении продуктов питания, содержащих фенилаланин. Если такая элиминационная диета применяется практически с рождения, человек вырастает здоровым.
Давайте узнаем поподробнее, что же это за заболевание, чем оно проявляется, как диагностируется и лечится.
Впервые описана в 1934 г. доктором Феллингом, откуда и получила свое второе название. Встречается с частотой в среднем 1: 10 000, но в разных странах мира существуют колебания от 1:2600 (в Турции) до 1:100 000 (в Финляндии и Японии), в России от 1:5 000 до 1:10 000.
Причины фенилкетонурии
В основе заболевания лежит генетический дефект – мутация гена 12-й хромосомы (98% всех случаев фенилкетонурии). Это так называемая классическая фенилкетонурия.
Ген кодирует количество фермента фенилаланин-4-гидроксилазы. Фермент отвечает за превращение аминокислоты фенилаланина в организме человека в тирозин в клетках печени.
Фенилаланин – это аминокислота, которая содержится в белковых продуктах (мясо, рыба, молоко, яйца и другие).
При мутации гена количество фермента снижается, что приводит к накоплению фенилаланина и продуктов промежуточного метаболизма в тканях организма. Организм пытается избавиться от фенилаланина и продуктов его распада, выводя их с мочой.Подобные нарушения обмена веществ приводят к нарушению строения нервных проводников, снижению образования нейромедиаторов. Все это, наряду с прямым токсическим действием избытка фенилаланина, приводит к развитию умственных нарушений, являющихся основным проявлением заболевания.
Фенилкетонурия наследуется по аутосомно-рецессивному типу, то есть не зависит от пола, и возникает при совпадении двух патологических генов от отца и матери.
Остальные 2% случаев фенилкетонурии связаны с другими генетическими дефектами и зависят от концентрации прочих ферментов (дигидроптеридинредуктазы и др.).
Они имеют те же клинические проявления, но не поддаются лечению диетой. Такие варианты относят к атипичному течению заболевания. Среди них принято выделять фенилкетонурию II и III.
Генетический дефект при фенилкетонурии II располагается в 4-й хромосоме, при III – в 11-й хромосоме.
Симптомы
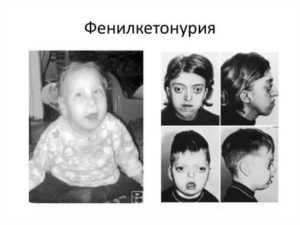
У детей с фенилкетонурией светлые волосы, бледная кожа и голубые глаза.
Ребенок с фенилкетонурией рождается внешне здоровым, то есть ничем не отличается от других детей. С поступлением пищи в организм начинается попадание белка, а значит и фенилаланина.
Последний постепенно накапливается, и обычно к 2 месяцам жизни появляются первые симптомы: вялость или беспокойство, отсутствие интереса к окружающему миру, срыгивания, изменения мышечного тонуса.
Иногда срыгивания столь частые и обильные, что возникает подозрение на патологию желудочно-кишечного тракта (пилоростеноз). Ребенок из-за срыгиваний может плохо набирать в весе.
К 4-6 месяцам становится очевидной задержка психического развития. Ребенок не следит за игрушкой, не реагирует на звук, не узнает родителей.Чем дольше продолжается поступление фенилаланина в организм с едой, тем выраженнее нарушения в психической и мыслительной сферах. Развитие речи резко задерживается. Иногда словарный запас может ограничиваться несколькими словами.
Если диагноз не будет выставлен и не будет начато лечение, то к 3-4 годам умственные нарушения достигнут степени идиотии (самая тяжелая степень олигофрении).
Особенностью клинического течения фенилкетонурии является необратимость возникших психических и интеллектуальных изменений. То есть при позднем выявлении помочь таким деткам уже нельзя – на всю жизнь они остаются умственно отсталыми.
Физическое развитие также отстает: дети позже начинают держать голову, переворачиваться, сидеть. Когда такие дети начинают ходить, то при этом они широко расставляют ножки, сгибая их одновременно в коленных и тазобедренных суставах.
Походка покачивающаяся, мелкими шажками. В положении сидя дети принимают «позу портного» — сгибают и руки, и ноги, поджимая последние под себя. Обычно объем головы меньше, чем в норме. Может быть выраженная микроцефалия: маленькая голова.
Из других неврологических симптомов возможны нарушения мышечного тонуса, судорожные припадки. Эпилептические приступы обычно появляются в возрасте 1,5 лет и приводят к еще большему прогрессированию нарушений интеллекта.
У части больных фенилкетонурией появляются непроизвольные движения в конечностях, дрожание (гиперкинезы). В движениях нет плавности и согласованности, нарушается равновесие.
Кроме ряда психических и интеллектуальных изменений, фенилкетонурию характеризуют следующие симптомы:
- специфический «мышиный» запах (или запах плесени) от ребенка: этот симптом характерен только для фенилкетонурии. Запах появляется в результате выделения продуктов метаболизма фенилаланина (фенилпировиноградной, фенилмолочной, фенилуксусной кислот) через кожу и с мочой;
- кожные проявления: дерматиты, экзема, просто шелушение (возникают по той же причине, что и «мышиный» запах);
- позднее прорезывание зубов: у таких детей первые зубы могут появиться после 18 месяцев, эмаль недоразвита;
- нарушение пигментации: у таких детей обычно голубые глаза, очень светлая кожа и волосы в результате снижения количества меланина (его содержание зависит от метаболизма фенилаланина). Из-за этого у таких детей наблюдается повышенная чувствительность к солнечному свету;
- вегетативные симптомы: пониженное артериальное давление, повышенная потливость, запоры, акроцианоз (синюшность кистей и стоп);
- нередко фенилкетонурия сопровождается врожденными пороками сердца.
Атипичные случаи фенилкетонурии, связанные с нарушением деятельности других ферментов, участвующих в метаболизме фенилаланина, кроме умственных изменений характеризуются развитием мышечной слабости во всех конечностях с одновременным повышением мышечного тонуса, спастическим тетрапарезом. Также при этих формах развивается слюнотечение, приступы повышения температуры.
У взрослых людей, страдающих фенилкетонурией, возможно появление судорожных припадков, нарушений координации, дрожания в конечностях, ухудшения памяти и внимания, возникновение депрессии. Обычно подобные симптомы возникают при несоблюдении элиминационной диеты.
Диагностика
Скрининговые тесты на содержание фенилаланина всем новорожденным проводят в роддоме.
В связи с тем, что фенилкетонурия сопровождается развитием необратимых умственных нарушений, во многих странах мира, в том числе и в России, принято использовать скриниг-методы диагностики.
Что это означает? Всем без исключения новорожденным детям в роддоме проводят экспресс-тесты на содержание фенилаланина.
Для этого берут капиллярную кровь (из пятки) на 4-5-й день жизни ребенка (у недоношенных на 7-й), наносят на специальный бумажный бланк и отправляют в лабораторию, где по определенным изменениям врач-лаборант делает выводы о содержании фенилаланина в крови. Отрицательный тест говорит об отсутствии фенилкетонурии.
Если тест оказывается положительным, то тогда проводят дополнительные исследования для определения содержания фенилаланина в крови и моче (хроматографию, флюориметрию). Концентрацию фенилаланина в крови и моче регулярно проверяют при проведении лечения, чтобы контролировать эффективность диеты и корригировать ее при необходимости.Возможно проведение генетического исследования для подтверждения мутации в гене, отвечающем за фенилаланин-4-гидроксилазу.
Подобное исследование возможно в качестве пренатальной диагностики, то есть на этапе беременности (берут околоплодные воды путем пункции).
Это инвазивное исследование делают по строгим показаниям (например, наличие больного фенилкетонурией ребенка в семье). Выявление генетического дефекта у плода позволяет прервать беременность.
Лечение фенилкетонурии
На сегодняшний день самым эффективным и распространенным способом лечения фенилкетонурии является элиминационная диета: диета с исключением продуктов, содержащих фенилаланин. Если ее строго придерживаться в первые годы жизни ребенка, когда развитие нервной системы еще продолжается, то можно вырастить здорового и полноценного человека.
Очень важно исключение фенилаланина именно в первый год жизни, когда наиболее активно развивается нервная система. Если элиминационная диета назначается после года, умственные нарушения не излечиваются. Каждый месяц первого года жизни без применения диеты обходится ребенку безвозвратной потерей около 4 баллов IQ.
Обычно достаточно придерживаться диеты до 16-18 лет, после этого возраста организм становится менее чувствительным к токсическому действию фенилаланина, и возможно расширение рациона питания. Включение новых продуктов необходимо проводить под контролем содержания фенилаланина в крови. Иногда требуется пожизненное строгое соблюдение диеты.
Беременным женщинам и женщинам, планирующим беременность, и при этом больным фенилкетонурией, для рождения здорового ребенка обязательно строгое соблюдение диеты.
Степень строгости диеты зависит от концентрации фенилаланина в крови у ребенка. При его уровне до 2-6 мг% (120-360 мкмоль/л) диета не назначается, выше этого показателя – обязательна.
Суть диеты заключается в исключении белковых продуктов.
Отказ от грудного вскармливания не обязателен, но в этом случае кормящая мать должна строго придерживаться элиминационной диеты, потому что грудное молоко содержит белок (соответственно и фенилаланин). Вопрос о возможности грудного вскармливания решается индивидуально!!!
Для пополнения запасов белка назначают специальные смеси, не содержащие фенилаланин – Афенилак, Лофеналак, Нофемикс. После года это Фенилфри, Нофелан, Бигрофен, Тетрафен, МД мил ФКУ-3 и другие.
В качестве прикорма назначают овощное и фруктовое пюре, фруктовые кисели, безбелковые каши (рисовая, кукурузная).После 6 месяцев можно применять специальные напитки Лопрофин, Нутриген и другие, кушать макаронные изделия, безбелковый хлеб.
В России обеспечение лечебным питанием детей, больных фенилкетонурией, по закону бесплатное.
Больным фенилкетонурией противопоказаны следующие продукты: мясо, рыба (и морепродукты), орехи, творог, твердый сыр, бобовые, яйца, изделия из пшеничной муки, гречневая и манная крупа, овсяные хлопья.
Во время назначения элиминационной диеты необходим строгий контроль содержания фенилаланина в крови: первые 3 месяца жизни – каждую неделю, от 3-х месяцев до года – минимум раз в месяц, от года до 3-х лет – 1 раз в 2 месяца. Стремятся фенилаланина 2-6 мг% у младших детей, после 10 лет – до 10 мг%. Обязательно наблюдение у детского психоневролога.
Кроме элиминационной диеты периодически назначаются комплексы из витаминов и минералов. Если есть судорожные припадки, необходимо применение антиконвульсантов (Депакин, Клоназепам и другие). Многим из таких детей показан массаж, лечебная физкультура. Возможно использование средств физиотерапии для коррекции мышечного тонуса.
Атипичные формы фенилкетонурии не поддаются лечению элиминационной диетой. В этом случае показано применение гепатопротекторов, антиконвульсантов, препаратов с Леводопой (для коррекции гиперкинезов), 5-окситриптофана, Тетрагидробиоптерина (ВН 4). Эти формы фенилкетонурии имеют худший прогноз для жизни и тем более интеллектуального развития.
На сегодняшний день разрабатываются новые направления в лечении фенилкетонурии. Среди них стоит отметить следующие:
- использование заместительной терапии фенилаланинлиазой (PAL) – растительным ферментом, расщепляющим фенилаланин до нетоксических соединений;
- генная инженерия (введение искусственно созданного нормального гена, ответственного за фенилаланин-4-гидроксилазу);
- метод «больших нейтральных аминокислот» — уменьшение всасывания фенилаланина из пищи и поступления в головной мозг с помощью специальных препаратов.
Пока эти современные разработки не имеют широкого применения, но некоторые исследования, подтверждающие их эффективность, уже проводятся.
Материнская фенилкетонурия
Если женщина, страдающая фенилкетонурией, при планировании беременности и при ее наступлении не придерживается соблюдения диеты, то это отражается на развитии ее ребенка. Потомство таких женщин имеет задержку внутриутробного развития и врожденные пороки развития: пороки сердца, аномалии развития головного мозга, мочевого пузыря, микроцефалию, аномалии лицевого скелета (расщелины).
Чтобы предотвратить патологические изменения у ребенка, таким женщинам необходимо придерживаться элиминационной диеты до зачатия и всю беременность. Дефицит белка восполняется за счет специальных белковых смесей без фенилаланина.
Таким образом, фенилкетонурия – это генетическое нарушение аминокислотного обмена, которое при поздней диагностике приводит к развитию выраженных интеллектуальных нарушений у ребенка.
Скрининг этого заболевания в роддомах позволяет диагностировать его в первые недели жизни и вовремя назначить лечение.Основным методом в настоящее время является назначение элиминационной диеты, которая позволяет сохранить интеллект маленькому человечку, а значит, сохранить здоровье, что обеспечит полноценное существование в течение всей жизни.
Источник: https://doctor-neurologist.ru/fenilketonuriya-simptomy-i-lechenie
Фенилкетонурия — что это, признаки, симптомы, лечение ~

Фенилкетонурия – это врожденное заболевание, передающееся генетически. Фенилкетонурия у детей связана с нарушением обмена веществ. Организм человека, больного этим недугом, не может расщеплять одну из аминокислот – фенилаланин, которая поступает в наше тело вместе с белковой пищей.
У здорового человека фениаланин расщепляется ферментами и становится тирозином. У человека с заболеванием фенилкетонурией нет фермента, расщепляющего фенилаланин.
Таким образом, эта аминокислота накапливается в физиологических жидкостях и тканях и приводит к поражению головного мозга и центральной нервной системы.
Данная болезнь была открыта в Норвегии доктором Иваром Фёллингом в 1934 году. Встречается фенилкетонурия у новорожденных довольно редко, в среднем 1 из 10 000 младенцев может иметь этот недуг. Хотя, в Турции и Словакии заболевание встречается намного чаще.
Фенилкетонурия. Признаки заболевания
Выглядеть новорожденный ребенок с фенилкетонурией может совершенно здоровым. Но позже, спустя пару месяцев, употребляя в пищу уже достаточно много пищи с фенилаланином, заметны первые симптомы.
Эта аминокислота содержится в белковой пище, а значит, что если мама кушает яйца, рыбу, гречку, сыр, орехи, и передает это ребенку через свое молоко, то, его организм, насытившийся фенилаланином, начинает отвергать его.
У малыша может наблюдаться:
— постоянное срыгивание, возможно даже срыгивание фонтаном после каждого приема пищи;
— потеря веса как следствие постоянного срыгивания;
— запах плесени от мочи ребенка;
— вялость и отсутствие интереса ко всему окружающему;
— слабый мышечный тонус;
— задержка в держании головки, переворотах на живот, хождении, речи, прорезывания зубов;
— нарушение концентрации, ребенок не слышит звуков, не смотри на родителей, не следит за предметами;
— пониженное артериальное давление, акроцианоз;
— возможны симптомы аллергического дерматита.
Все это — симптомы фенилкетонурии.
Фенилкетонурия у человека зачастую сопровождается приступами эпилепсии. Если вовремя не обратиться к врачу, то к трем годам у ребенка может развиться олигофрения и даже идиотия.
Диагностика фенилкетонурии
В настоящее время в роддоме всем новорожденным проводят скрининг-тесты на содержание фенилаланина. Для этого, у каждого младенца приблизительно на 4-ый день жизни из пяточки берут кровь. Далее, нанеся кровь на специальный бланк, образец отправляют в лабораторию.
Если лаборант вынесет отрицательный вердикт, значит фенилкетонурия у младенца отсутствует. Если же результат окажется положительным, проводятся дополнительные манипуляции. Специалисты проводят хроматографию и флюориметрию.
Если беременная женщина очень волнуется за возможность появления такой болезни у своего ребенка вследствие наличия ее у отца или матери, можно провести этот скрининг-тест посредством взятия пункции из околоплодных вод матери.
Что такое фенилкетонурия
Фенилкетонурия (ФКУ) – это аутосомно-рецессивное генетическое заболевание, связанное с нарушением метаболизма аминокислот.
Фенилкетонурия как самостоятельное заболевание впервые было описано в 1934 году врачом из Норвегии по имени Ивар Асбьёрн Фёллинг. В то время заболевание получило название гиперфениллалниемия, основным симптомом которого была именно задержка умственного развития. В Норвегии это заболевание до сих пор известно как «болезнь Фёлинга».
А вот успешное лечение заболевания впервые было проведено в Англии в Бирмингемском детском госпитале. Произошло это событие в 50-х годах XX века. Лечение проводила целая группа медиков, руководил которыми Хорста Биккеля.
Окончательно лечить и диагностировать заболевание научились только в начале 60-х годов XX века. Именно в это время был разработан специальный тест, который помогал в первые несколько дней после рождения ребёнка по анализу крови выявить это серьёзное заболевание. Назывался тест метод Гатри.
Со временем о заболевании стало известно всё больше и больше, и диагностировать и лечить его научились уже с самого первого дня выявления. Да и сама диагностика сегодня не представляет какой-либо проблемы.
Фенилкетонурия является наиболее распространённым наследственным нарушением аминокислотного обмена. Частота данной патологии среди новорождённых, по данным массового обследования, в различных странах сильно варьирует: от 1:4000 в Ирландии и Турции до 1:80000 в Японии. В Республике Беларусь частота фенилкетонурии составляет 1:6000 новорожденных .
В связи с достаточно высокой распространённостью в популяции, тяжестью клинических проявлений и возможностью профилактического лечения, фенилкетонурия в числе первых наследственных нарушений обмена веществ была включена в список наследственных заболеваний, рекомендованных Всемирной организацией здравоохранения для раннего выявления у новорожденных.
Мутация R408W является наиболее распространенной у белорусских пациентов с ФКУ (67%).
Поскольку ФКУ наследуется по аутосомно-рецессивному типу, риск повторения для последующих детей в семье, где есть больной ребенок, составляет 25 % и не зависит ни от числа больных детей в семье, ни от их пола.
Что происходит в организме при фенилкетонурии?
— Любой белок, поступающий в организм с пищей, состоит из аминокислот. Фенилаланин ─ одна из аминокислот. В организме здорового человека фенилаланин трансформируется в тирозин, это тоже аминокислота.
А тирозин участвует в синтезе гормонов, пигментов, нейромедиаторов ─ веществ, которые нужны для нормальной деятельности центральной нервной системы.
Превращению фенилаланина в тирозин способствует фермент фенилаланингидроксилаза, который кодирует одноименный ген.
Если в гене есть дефект, он есть и в ферменте. Мутированный фермент не выполняет свою функцию. В результате фенилаланин не превращается в тирозин, тирозина организму не хватает, а фенилаланин, наоборот, накапливается и начинает распадаться.
Аминокислота и продукты ее распада ─ фенилпировиноградная, фенилмолочная и фенилуксусная кислоты и другие, в избытке действуют на организм, как яд.
Симптомы фенилкетонурии
Первые признаки проявляются в возрасте от 2 до 6 месяцев и имеют неспецифический характер: по ним нельзя сказать, фенилкетонурия это или нет. Малыш может быть вялым, раздражительным, беспокойным, часто плачет.
С такой неврологической симптоматикой педиатр может поставить диагноз «внутричерепное давление». Наблюдаются высыпания на коже ─ будет поставлен диагноз «диатез».
При отсутствии лечения вскоре к первым симптомам присоединяется специфический мышиный запах мочи, пота ─ так называемый запах грязных пеленок. Он говорит о том, что в организме ребенка в большом количестве скопились ядовитые продукты обмена веществ.В дальнейшем у больного развиваются судорожный синдром, который не поддается обычной терапии судорог, поведенческие расстройства тяжелой степени, умственная отсталость крайней степени ─ идиотия.
Основными проявлениями патологии служат:
- расстройство интеллектуального развития
- экзема
- вялость
- судороги
- мышечный тремор
- эпилепсия
- «мышиный» запах
Диагностика заболевания
Обычно фенилкетонурию определяют в процессе скрининга новорождённых.Для выявления ФКУ используют высокоэффективную жидкостную хроматографию.
В Беларуси с 1978 года обследованию на ФКУ подлежат все новорожденные дети: доношенные на 3-й день, недоношенные на 7-14-е сутки жизни.В условиях нашей страны лечение вводится в среднем на 22-й день жизни ребенка.
В связи с массовым характером обследования новорожденных на ФКУ в Белоруссии нелеченые формы заболевания могут встретиться лишь:
1) у детей, родившихся вне пределов республики;
2) у детей, которым по каким-то причинам не было сделано обследование в послеродовом периоде (по разным причинам обследовать удаётся практически лишь 93—95 % всех новорожденных).
Тестирование проводится в клинико-диагностической генетической лаборатории Республиканского научно-практического центра (РНПЦ) «Мать и дитя» в сухих пробах крови. Скринирующим тестом на ФКУ является определение ФА флюорометрическим методом. Границей нормальных значений ФА является концентрация 180 мкмоль/л.
При превышении этих значений осуществляется повторное тестирование, которое должно быть проведено до двухнедельного возраста ребенка.
При сохранении повышенного уровня ФА рекомендуется в трехдневный срок госпитализировать ребенка в РНПЦ «Мать и дитя» для верификации диагноза, начала терапии и консультации врача-генетика.
Как лечить?
Единственный (доступный на сегодняшний день для большинства) метод лечения заключается в своевременной организации диетотерапии, которая требуется с первых дней жизни. Заключается она в резком ограничении фенилаланина и возмещении белка за счет аминокислотных смесей.
В Республике Беларусь бесплатное обеспечение специализированными смесями проводится до 18 лет. В последнее время установлено, что головной мозг страдает от избытка ФА на протяжении всей жизни, поэтому диету рекомендуют соблюдать всю оставшуюся жизнь.
Потребность организма в ФА зависит не только от возраста и активности, но и от индивидуальной переносимости, обусловленной степенью остаточной активности фермента ФАГ.
Однако необходимо помнить, что недостаточное потребление ФА также опасно, как и избыточное.Ведение лечения:
1. Дети с ФКУ до диетотерапии госпитализируются в инфекционное отделение новорожденных РНПЦ «Мать и дитя», где наблюдаются врачом-генетиком, врачом-педиатром и (по показаниям) врачом-невропатологом.
2. Ведение диетотерапии смесью аминокислот осуществляют в течение 6–7 дней: ежедневно одно кормление заменяется смесью аминокислот (под контролем ФА)
3. В дальнейшем родителей ребенка обучают расчету питания в зависимости от суточной потребности ФА и массы тела.
Возможно ли кормление грудью?
При фенилкетонурии допустимо кормление грудью. Надо лишь контролировать с помощью анализов уровень фенилаланина в крови больного ребенка.
Самой же кормящей матери ограничивать потребление белков не надо, так как их количество в грудном молоке практически неизменно, независимо от продуктов питания матери.
Более того, кормление грудью больных фенилкетонурией детей очень ценно, и если есть хоть малейшая возможность кормить такого ребенка грудным молоком, эту возможность упускать нельзя.
Типы фенилкетонурии:
Классическая фенилкетонурия– фа > 1200 ммол/л (20 мг%)
Умеренная фенилкетонурия – фа – 900 -1200 ммол/л (15 — 20 мг%)
Мягкая фенилкетонурия – фа –600–900 ммол/л (10-15мг%)
Мягкая гиперфенилаланинемия – фа -360 – 600 ммол/л (6 — 10мг%)
Мягкая гиперфенилаланинемия— не нуждающаяся в лечении – фа – 120 – 360 ммол/л (2 — 6мг%)
Злокачественная гиперфенилаланинемия – недостаточность тетpагидpобиоптеpина (BH4)
Что такое «переносимость»
Переносимость-это то количество белка(=фенилаланина),которое человек с фку может употребить за сутки,и при этом иметь анализы в пределах нормы.Переносимость ребёнка становится известной к 2-3 годам.
Источник: http://fenilka.by/%D1%87%D1%82%D0%BE-%D1%82%D0%B0%D0%BA%D0%BE%D0%B5-%D1%84%D0%B5%D0%BD%D0%B8%D0%BB%D0%BA%D0%B5%D1%82%D0%BE%D0%BD%D1%83%D1%80%D0%B8%D1%8F/
Фенилкетонурия — Генетик-РО

проф. Круглов Сергей Владимирович (слева), Крючкова Оксана Александровна (справа)
Автор проекта: Круглов Сергей Владимирович, профессор, заслуженный врач России, доктор медицинских наук, врач высшей квалификационной категории
Подробнее
Редактор страницы: Крючкова Оксана Александровна — врач-травматолог-ортопед
Подробнее
Амелина Светлана Сергеевна — профессор кафедры по курсу генетики и лабораторной генетики, доктор медицинских наук. Врач генетик высшей квалификационной категории
Прочитать о докторе подробнее
Дегтерева Елена Валентиновна — ассистент кафедры по курсу генетики и лабораторной генетики, врач-генетик первой категории
Прочитать о докторе подробнее
Фенилкетонурия – синоним – Феллинга болезнь или олигофрения фенилпировиноградная. Это заболевание, которое наблюдается у детей сразу после рождения. Поэтому относится к группе врожденных и генетически обусловленных.
Развивается данная патология вследствие того, что в организме родившегося ребенка наблюдается нарушение к восприимчивости и переработки фенилаланина, поступающего с пищей.
Вследствие данного нарушения, при несвоевременной диагностике наблюдается поражение структур головного мозга ребенка и с последующим развитием умственной отсталости (при условии отсутствия должного лечения).
Фенилкетонурия: эпидемиология
Фенилкетонурия
Данные о этой патологии впервые появились еще в 1934 году, ученый, который впервые предоставил данные о заболевании – А.Феллинг. частота встречаемости данной патологии составляет 1:10000 младенцев.
В раннем послеродовом периоде никаких проявлений данного заболевания у новорожденного нет. Если данное заболевание не лечить, то ы течение первого года жизни ребенка данная патология вызывает необратимые нарушения в развитии центральной нервной системы, вызывая необратимые изменения.
Именно поэтому диагностические мероприятия, направленные на выявление данной патологии необходимо проводить сразу после рождения ребенка, еще на этапе родильного дома, так как ранняя диагностика играет решающее значение в профилактике тяжелых последствий фенилкетонурии.
Фенилкетонурия: причины развития
Данная патология относится к заболеваниям, наследующимся по аутосомно-рецессивному типу. Расшифровывается данное выражение так: чтобы развились клинические проявления фенилкетонурии у младенца, оба родителя должны быть носителями поврежденного гена, отвечающего за данную патологию.
Поврежденный ген, ответственный за развитие фенилкетонурии располагается в 12 хромосоме, и отвечает за то, чтобы присутствовал фермент фенилаланин – 4 гидролаза. Если повреждается именно данная хромосома, то происходит развитие у ребенка классического типа фенилкетонурии – 1 типа.
Данный тип подобной патологии в процентном соотношении составляет практически девяносто восемь процентов от всех случаев заболевания.
При развитии фенилкетонурии 1 типа – числовые значения фенилаланина в организме младенца достигает 30 мг%, иногда и более. Если не начать лечебные мероприятия максимально рано – то ребенок в итоге становится умственно отсталым. Причем умственная отсталость выражена в самой крайней степени.
Также у новорожденного может развиваться и атипичная форма фенилкетонурии. В чем же атипичность данных форм? Атипичные формы данной патологии невозможно корректировать диетотерапией!.
Фенилкетонурия: атипичные формы:
1. II тип фенилкетонурии. При данном патологии наблюдается недостаточность такого фермента, как дегидроптеринредуктаза.
2. III тип фенилкетонурии. Приданном варианте патологии наблюдается недостаточность тетрагидробиоптерина.
Помимо двух ранее перечисленных варианта атипичной формы фенилкетонурии, существуют и иные варианты, но они практически не встречаются в практике врача.Необходимо также понимать, что если родители находятся в близкородственном браке (часто встречается среди некоторых национальностей), то повышается вероятность того, что ребенок может родится с фенилкетонурией.
Фенилкетонурия: механизм развития данной патологии
Основная причина развития классической формы фенилкетонурии 1 типа, как уже было сказано, является дефицит такого фермента, как фенилаланин – 4 – гидролазы. Этот фермент необходим для того, чтобы фенилаланин переходил в следующую фракцию – тирозин. Это происходит в клетках печени.
После того, как тирозин образовался, он, а точнее его производные, необходим для того, чтобы образовались из него в свою очередь гормоны надпочечников и щитовидной железы (адреналин, норадреналин и тироксин соответственно).
Помимо того, что фенилаланин необходим для формирования в организме ребенка гормонов, он еще участвует в формировании меланина – вещества, которое отвечает за формирование пигментации.
Если у ребенка имеется нарушение в ферментативной составляющей, то переработка поступающего, с пищей, в организм ребенка, фенилаланина нарушается, а, следовательно, содержание фенилаланина в крови и ликворе возрастает в геометрической прогрессии, при этом количественное содержание тирозина снижается. Избыток начинает выводится через почки вместе с мочой. Причем с мочой ребенка выделяется не сам фенилаланин, а его производные – фенилпировиноградная, фенилмолочная или фенилуксусная кислоты.
Итак, мы пришли к тому, что в организме ребенка происходит катастрофа. Выражается она в том, что аминокислотный обмен нарушен, а данное нарушение напрямую отражается на нервной системе (нарушение обмена аминокислот — нарушение образования миелиновых волокон в головном мозге ребенка/ нейромедиаторов вырабатываются в меньшем количестве – задержка умственного развития – слабоумие).
Фенилкетонурия: клиническая картина
1. Разгар заболевания приходится на возраст от двух до шести месяцев. Связано это с тем, что при кормлении младенца, к нему в организм начинает поступать белок, который и является катализатором, приводящим к развитию симптомов.
2. Вялость или обратная сторона – беспокойство/гипервозбудимость.
3. Срыгивание
4. Синдром судорожный
5. Дистония мышечная
6. Специфичный симптом – рвота, которая приобретает упорный характер. Иногда при недостаточной диагностике врач ставят ошибочный диагноз – пилоростеноз или пилороспазм.
7. После шести месяцев от момента рождения малыша становится ярко видно, что он отстает в нервно-психическом развитии от остальных детей. — малыш неактивный, не узнает родителей, не садится, не пытается ходить.
8. Специфичный признак – запах плесени («мышиный»), который исходит от тела новорожденного. Это связано с тем, что с мочой и с потом выходят непереработанные фракции фенилаланина.9. Шелушение кожных покровов, и как следствие дерматит/экзема/склеродермия.
10. Малый размер черепа. Можно заметить в том случае, если малышу не проводилось никакого лечения по поводу фенилкетонурии.
11. Нарушение зубной формулы, прорезывания зубов, их вида. Становится заметно лишь в полуторогодовалом возрасте.
12. Помимо этого, к 1 году становится заметна задержка речевого развития, если на данном этапе не проводить никакого лечения, то к трем годам малыш совсем не может говорить, а врачи выставляют диагноз – умственная отсталость – олигофрения.
13. Телосложение у малышей с подобной патологией непропорциональное. Кожные покровы, цвет глаз и волос – светлые.
14. Возможны пороки сердечно-сосудистой системы.
15. Дисфункция со стороны вегетативной нервной системы – малыш много потеет, ручки и ножки бледные с синеватым оттенком, давление ниже нормальных, для ребенка, величин.
16. Характерно – поза «портного» — ручки и ножки малыша согнуты в суставах, дрожат. Если малыш ходит- то следует обратить внимание на походку ребенка – она шаткая, семенящая. Также можно наблюдать гиперкинезы.
Это были описаны признаки, которые характерны для так называемой классической фенилкетонурии.
Разберем же, признаки специфические для аномальных форм. Итак:
1. II тип фенилкетонурии – умственная отсталость проявляется в самой тяжелой форме – олигофрении. Малыш возбудим, отмечаются судороги, спастический тетрапарез, сухожильные рефлексы выражены чрезмерно. Вследствие того, что заболевание прогрессирует, то приводит к смерти ребенка в возрасте 3 лет.
2. III тип фенилкетонурии. Для данного типа характерна триада: малый размер черепа и головного мозга, умственная отсталость в виде олигофрении, тетрапарез спастический.
Фенилкетонурия: диагностические мероприятия
Самый главный этап диагностики – это скрининг в неонатальном периоде.
После родов всем новорожденным на третий – пятый день (седьмой день – если ребенок родился недоношенным) от момента родов, осуществляется забор крови для диагностики данной патологии (а также для диагностики галактоземии, гипотиреоза врожденного, синдрома адреногенитального). При получении результатов обследования –гиперфенилаланемия от 2,2мг% или более – это повод для того, чтобы ребенка отправить на консультацию к генетику.
Врач-генетик для верификации диагноза фенилкетонурии назначает сдать анализ крови (определяют уровень фенилаланина, тирозина, ферментов печени), анализ мочи (определяют количество кислот кетоновых, производных катехоламина).
Кроме всего ранее перечисленного выполняют электроэнцефалограмму, магнитно-резонансную томографию головного мозга. После проведения данных исследований рекомендована консультация невролога детского, для уточнения наличия или отсутствия поражения структур головного мозга.Если фенилкетонурия является диагнозом уже на этапе беременности, то женщине показано выполнение диагностических мероприятий – амниоцентеза, кордоцентеза, хорионбиопсии.
Фенилкетонурия: дифференциальный диагноз
Как уже было сказано ранее, то данную патологию необходимо дифференцировать с рядом заболеваний, которые не имеют отношения к фенилкетонурии. Итак:
1. Пилоростеноз
2. Пилороспазм
3. Родовая травма внутричерепная (кровоизлияния и т.д.)
4. Инфекционные поражения внутриутробного характера.
5. Нарушения обмена аминокислот, не связанные с фенилкетонурией.
Фенилкетонурия: лечебные мероприятия
1. Первое и основное – диета. Поступление белка в организм малыша должно быть ограничено. Рекомендована в том случае, когда целевые значения фенилаланина составляют уже более шести мг%. На данный момент разработаны ряд искусственный смесей для детей, страдающих фенилкетонурией.
2. Показаны детям с фенилкетонурией фрукты, овощи, соки, гидролизаты белка, смеси содержащие аминокислоты. Расширять диету можно лишь после того, как ребенку исполнится 18 лет, так как в данном возрасте толерантность к фенилаланину возрастает.
3. Прием соединений минеральных, витаминных комплексов
4. Средства, улучшающие кровоснабжение головного мозга, снижающие частоту судорог
5. Массаж
6. Лечебная физкультура
7. Наблюдение врача-педиатра, невролога
8. Постоянный контроль за неврологическим состоянием пациента. Регулярно выполняют электроэнцефалограмму.
9. Постоянный контроль показателей крови.
Фенилкетонурия: профилактика
1. Максимально ранняя диагностика фенилкетонурии. Чем раньше диагностировано заболевание, тем меньшие осложнения наблюдаются у ребенка. Для ранней диагностики выполняется скрининг новорожденных.
2. Генетическое консультирование. Необходимо для тех семей, где брак является кровнородственным, или же уже имеется ребенок, страдающий данным заболеванием. Необходимо помнить, что если женщина больна фенилкетонурией, то на всех этапах – планировании беременности, вынашивании плода, она должна соблюдать диету!.
Необходимо понимать, что все погрешности, которые допускает будущая мать напрямую отражаются на ребенке, которого она носит. Ведь если родители не больны, но являются носителями «поломанного» гена, то риск того, что малыш родится больным уже равен двадцати (!) процентам.Проще говоря из 4 беременностей, 1 закончится рождением ребенка с фенилкетонурией.
Запись на прием к генетику:
Уважаемые пациенты, Мы предоставляем возможность записаться напрямую на прием к доктору, к которому вы хотите попасть на консультацию. Позвоните по номеру ,указанному вверху сайта, вы получите ответы на все вопросы. Предварительно, рекомендуем Вам изучить раздел О Нас.
Как записаться на консультацию врача?
1) Позвонить по номеру 8-863-322-03-16.
2) Вам ответит дежурный врач.
3) Расскажите о том, что вас беспокоит. Будьте готовы, что доктор попросит Вас рассказать максимально подробно о своих жалобах с целью определения специалиста, требующегося для консультации. Под руками держите все имеющиеся анализы, особенно, недавно сделанные!
4) Вас свяжут с вашим будущим лечащим доктором (профессором, доктором, кандидатом медицинских наук). Далее, непосредственно с ним вы будете обговаривать место и дату консультации — с тем человеком, кто и будет Вас лечить.
ОК: 19.01.2017г.
Источник: http://genetic-rost.ru/%D0%B8%D0%BD%D1%84%D0%BE%D1%80%D0%BC%D0%B0%D1%86%D0%B8%D1%8F/%D1%84%D0%B5%D0%BD%D0%B8%D0%BB%D0%BA%D0%B5%D1%82%D0%BE%D0%BD%D1%83%D1%80%D0%B8%D1%8F/