Мозаичная трисомия 22 хромосомы
Трисомия 9, несколько вариантов

Синдром трисомии 9 хромосомы мозаичной формы – редкое хромосомное расстройство, при котором часть 9-й хромосомы появляется три раза, а не дважды в клетках тела. Термин «мозаика» указывает, что некоторые клетки содержат дополнительную хромосому.
Короткое плечо хромосомы 9 включает в себя полосы 9p11 – 9p24, а длинное полосы 9q11 – 9q34.
Во многих случаях симптомы схожи среди пораженных детей, несмотря на разную длину дублированного сегмента. Однако у пациентов с более крупными трисомами (например, простирающимися до средних или конечных областей 9q) могут присутствовать дополнительные признаки.
Кроме того, некоторые люди с дублированием конкретных областей хромосомы 9p не развивали никаких симптомов или только очень мягкие.
У детей с синдромом трисомии хромосомы 9 присутствуют задержки этапов развития, включая ходьбу, дефицит роста, отличительные пороки развития черепа и лицевой (черепно-лицевой) области. По мере взросления умственная инвалидность становится очевидной.
В некоторых случаях могут присутствовать дополнительные физические аномалии, такие как скелетные дефекты, структурные пороки развития сердца (врожденные).
Расстройство является результатом сбалансированной хромосомной перегруппировки у одного из родителей или возникает из спонтанных ошибок в эмбриональном развитии, которые происходят по неизвестным причинам.Первоначально хромосомная аномалия была зарегистрирована в медицинской литературе в 1970 году. Трисомия 9p впервые предложена как особый синдром с характерными симптомами в 1975 году.
Симптомы
Конкретные симптомы синдрома трисомии 9 хромосомы мозаичной формы варьируются от одного человека к другому. Они зависят от определенной длины дублированного генного материала.
Trisomy 9p характеризуется:
Это означает, что скорость роста и развития костей медленнее, чем у человека с 46 хромосомами. Гипотония поражает младенцев, чаще с трудностями кормления, что приводит к неспособности набрать вес и расти с ожидаемой скоростью.
Дефицит роста начинается после рождения (постнатально). У некоторых младенцев может наблюдаться орофарингеальная дисфагия, при которой трудно продвигать пищу из задней части горла (орофаринкс) в пищевод.
У людей с более крупными трисомными сегментами (например, полосы 9q22 или 9q32) дефицит роста начинается до рождения (внутриутробная задержка роста).
Во многих случаях синдром трисомии хромосомы 9 связан с разной степенью умственной неполноценности от умеренной до тяжелой, задержками приобретения навыков, требующих координации умственной и физической деятельности (расстройства координации развития).
Наиболее сильно проявляется задержка развития речи. Наблюдаются инвалидности, начиная от легкой до тяжелой. Интеллектуальные и учебные проблемы идут рука об руку. Интеллектуальные проблемы имеют широкий диапазон, особенно у детей с частичной трисомией 9. Разрыв с типично развивающимися сверстниками часто увеличивается с возрастом.
Внешний вид
Многие младенцы и дети с трисомией 9 имеют характерный внешний вид лица. Большинство людей с этим состоянием имеют:
- короткую и широкую голову (брахицефалию);
- широкий рот с крутыми углами;
- видный, луковичный нос;
- большие, низкорослые, «чашевидные» уши;
- короткую вертикальную канавку в центре верхней губы (philtrum).
Также присутствуют характерные аномалии глаз, такие как:
- глубоко посаженные, широко расставленные глаза;
- наклонные веки (пальпебральные трещины);
- вертикальные складки кожи, которые покрывают внутренние углы глаз (эпикантальные складки);
- аномальное отклонение одного глаза по отношению к другому (косоглазие).
У некоторых затронутых младенцев может быть короткая шея; высокая арочная крыша рта (неба). Зубы прорезываются позже, чем ожидалось, растут криво.
Узнать больше Болезнь Альперса – симптомы, лечение
Опорно-двигательная система
У многих присутствуют различные аномалии рук и ног:
- уменьшенную длину отдельных костей пальцев (фаланг), пястных костей, стопы (метатарзалы);
- короткие пальцы с маленькими ногтями;
- мизинцы бывают зафиксированы в изогнутом положении (клинодактильно).
Для синдрома характерны необычные, отличительные узоры кожи рук (аномальные дерматоглифы), уменьшенное количество пальцев. Менее часто присутствует одна складка на ладонях.
Расстройство связано со скелетными дефектами:
- задержка закрытия родничков и фиброзных суставов (черепных швов) костей черепа;
- деформация, при которой нога выкручивается (косолапость);
- ненормальная передняя, задняя, боковая кривизна позвоночника (кифосколиоз), развивающийся во втором десятилетии жизни.
Встречается частичное сращивание (синдактилия) пальцев, вывих бедер при рождении.
Сердечно сосудистая система
От 5 до 25% детей имеют врожденные пороки сердца, аномальное открытие перегородки, которая разделяет две нижние камеры (желудочки) сердца (дефекты желудочковой перегородки [VSD]).
Симптомы варьируются в зависимости от размера, характера или комбинации пороков развития сердца. Например, с маленькими изолированными VSD, симптомы бывают не очевидными (бессимптомными).
Дети с более крупными пороками VSD или другими сердечными дефектами, демонстрируют трудности при кормлении, одышку, обильное потоотделение, раздражительность, усталость, синеватый окрас кожи или слизистых оболочек (цианоз).При тяжелых случаях, врожденная болезнь сердца может привести к потенциально опасным для жизни осложнениям. Требуется хирургическое вмешательство для устранения сердечных дефектов с последующим наблюдением.
Дополнительные признаки
Сообщалось о дополнительных физических аномалиях. К ним относятся:
- генитальные пороки развития у пораженных мужчин, такие как неопущенные семенники (крипторхизм), аномальное размещение мочевого отверстия (гипоспадии);
- почечные пороки развития;
- протрузия части кишечника, отложение жировой мембраны перед кишечником (сальник) через дефект в брюшной стенке пупка (пупочная грыжа);
- гидроцефалия, при которой блокирование нормального потока спинномозговой жидкости приводит к ее избытку, накапливающемуся в мозге и вокруг него. Это приводит к аномально высокому черепному давлению, отеку, неврологическим нарушениям.
В зависимости от возраста возникновения симптомов и других факторов, возникают внезапные эпизоды неконтролируемой электрической активности головного мозга (судороги), раздражительность, рвота, головная боль, потеря координации, ухудшение психического функционирования.
Некоторые индивидуумы развивают мальформацию Дэнди-Уокера (DWM). DWM происходит во время эмбрионального развития мозжечка и 4-го желудочка.
Мальформация Дэнди-Уокера характеризуется неправильным развитием (малым размером, аномальным положением) средней части мозжечка, кистозным расширением 4-го желудочка и основания черепа. Связан с гидроцефалией.
Пострадавшие дети имеют рост ниже среднего для своего возраста. Иногда присутствует дефицита гормона роста.
Причины
Синдром трисомии 9 хромосомы возникает из-за спонтанных ошибок при эмбриональном развитии, происходящих по неизвестным причинам. В таких случаях родители затронутого ребенка имеют нормальные хромосомы и низкий риск рождения другого с хромосомными аномалиями.
Примерно в 50% случаев трисомия 9 вызвана сбалансированной хромосомной перестройкой одного из родителей. Родительская перегруппировка описывается как «сбалансированная транслокация».
Транслокации происходят, когда фрагменты прерываются, перегруппируются, что приводит к смещению генетического материала и измененному набору хромосом. Однако генетический материал не утрачивается, а только перегруппируется.
Если хромосомная перегруппировка сбалансирована, обычно безвредна для носителя, но связана с повышенным риском развития аномальности у потомков, например, трисомия х.Сообщалось о редких случаях, когда исходная хромосомная перегруппировка была инверсией. Инверсия характеризуется поломкой хромосомы в двух местах и воссоединением сегмента в обратном порядке от первоначальной компоновки.
Хромосомный анализ и генетическое консультирование рекомендуются родителям пострадавшего ребенка, чтобы подтвердить или исключить наличие сбалансированной транслокации или другой хромосомной перегруппировки с участием хромосомы 9 у одного из родителей.
Узнать больше Что такое токсический шок?
Специфика
Исследователи считают, что 9p22 является «критическим» регионом, ответственным за большинство классических симптомов трисомии 9.
Затронутые популяции
Влияет на женщин в два раза чаще, чем мужчин. Синдром трисомии 9 хромосомы является четвертым по распространенности после трисомии 21 (синдром Дауна), 18 (синдром Эдвардса), 13 (синдром Патау).
Связанные нарушения
Симптомы следующих расстройств могут быть сходными. Сравнения полезны для дифференциального диагноза:
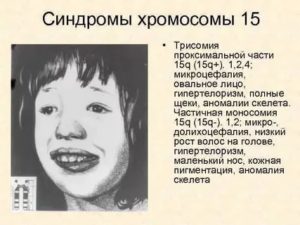
Тетрасомия 9
Редкое генетическое расстройство, где короткое плечо хромосомы присутствует четыре раза (тетрасомия), а не дважды во всех или некоторых клетках организма. Многие симптомы похожими на те, которые наблюдаются у людей с трисомией. Характерные аномалии, связанные с тетрасомией 9, включают:
- дефицит роста;
- нарушение развития;
- умственную неполноценность от умеренной до тяжелой;
- различные черепно-лицевые, скелетные, сердечные, почечные и / другие физические дефекты.
Краниофациальные аномалии похожи и имеют:
- луковичный нос;
- уродливые уши;
- наклонный рот;
- глубоко укоренившиеся, широко расставленные глаза;
- короткую шею.
Микроцефалия проявляется в младенчестве, окружность головы меньше, чем ожидаемая, исходя из возраста и пола младенца. Тетрасомия 9 вызвана спонтанными ошибками в самом начале эмбрионального развития, которые происходят случайным образом по неизвестным причинам.
Мозаичная форма
Расстройство, характеризующееся трисомой всей 9-й хромосомы в некоторых клетках тела (мозаицизм). Термин «мозаицизм» указывает, что некоторый процент клеток пострадавшего человека имеет хромосомную аномалию.
Диапазон и тяжесть симптомов зависят от процента клеток с дополнительным материалом. Характерные особенности включают:
- дефицит роста до рождения (внутриутробная замедление роста);
- врожденные пороки сердца;
- скелетные, генитальные, почечные, неврологические аномалии;
- переменные черепно-лицевые дефекты, такие как луковичный нос, короткие складчатые веки (пальпебральные трещины), глубоко посаженные глаза, небольшая челюсть (микрогнатия), низкорослые и уродливые уши, короткая шея.
Мутация вызвана ошибками во время деления репродуктивных клеток (мейоза) родителя или во время деления клеток ткани тела (соматических клеток) на ранних стадиях развития эмбриона (митоза). Причина таких ошибок неизвестна.
Диагностика
Часто диагноз устанавливается до рождения (пренатально) с помощью специализированных тестов, таких как ультразвук, амниоцентез, хорионная выборка ворсинок (CVS).
Генетические исследования, проведенные на образцах жидкости или ткани, выявляют трисомию части или всего короткого плеча хромосомы 9 (9р), иногда, части длинного плеча (9q).
Trisomy 9p также диагностируется, подтверждается после рождения путем тщательной клинической оценки, идентификации характерных физических признаков, генетического анализа, других специализированных тестов.
Лечение
Лечение синдрома трисомии 9 хромосомы направлено на конкретные симптомы, физические признаки. Оно требует скоординированных усилий группы медицинских специалистов, таких как:
- педиатры;
- хирурги;
- врачи, лечащие аномалии скелета, суставов, мышц (ортопеды);
- кардиологи;
- неврологи.
Генетическая консультация рекомендуется для семей с детьми, пострадавшими от этого состояния. Возможно, потребуется психосоциальная поддержка всей семьи.
Диагностика во время младенчества и детства (до трех лет) имеет важное значение для того, чтобы дети достигли своего потенциала. Очень важна ранняя речевая терапия. Дополнительные услуги включают специальное образование, физическую, профессиональную терапию, логопедию.
Индивидуальная образовательная программа должна быть разработана для оказания помощи детям в школе, чтобы ребенок получил доступ к равному образованию. Реабилитационная поддержка часто необходима во взрослой жизни.
Дополнительное лечение расстройства является симптоматическим. Например, для врожденных пороков сердца может потребоваться лечение лекарственными средствами, хирургическое вмешательство (паллиативное, корректирующее).
Врачи могут рекомендовать хирургическую коррекцию характерных черепно-лицевых мальформаций, скелетных аномалий, генитальных дефектов, грыж, аномалий почек, других пороков развития.
Конкретные хирургические процедуры зависят от характера и тяжести анатомических аномалий, связанных с ними симптомов, других факторов. Дефицит гормона роста успешно лечится при помощи гормональной терапии.
Источник: https://ovp1.ru/genetic/trisomiya-9-mozaichnoj-formy
Самые частые патологии плода во время беременности. Обзор синдромов

Пренатальная диагностика — это комплекс генетических исследований во время беременности, проводящийся с целью обнаружения патологии у плода на стадии внутриутробного развития.
Данный вид исследований позволяет обнаружить более 99 % плодов с синдромом Дауна (или трисомия по 21 хромосоме), синдромом Эдвардса (трисомия по 18 хромосоме),синдромом Патау (трисомия по 13 хромосоме). Хромосомные аномалии имеют место в 1 из 150 случаев деторождения.
В случае наличия у плода болезни родители при помощи врача-генетика тщательно взвешивают возможности современной медицины и свои собственные в плане реабилитации ребёнка.
В результате семья принимает решение о судьбе данного ребёнка и решает вопрос о продолжении вынашивания
Пренатальная диагностика подразделяется на инвазивную и неинвазивную
- Инвазивные методы диагностики – такие как биопсия хориона, амниоцентез (забор околоплодных вод), кордоцентез (забор крови из пуповины).
- Неинвазивные методы диагностики более щадящие. К ним относят неинвазивный пренатальный тест на частые хромосомные аномалии, которые совместимы с жизнью, но такие хромосомные аномалии как синдромы Дауна, Эдвардса и Патау сопровождаются множественными пороками развития и не поддаются излечению. НИПТ надежнее биохимического скрининга, так как относится к прямым методам исследования плодной ДНК. При этом, у будущей мамы нет риска напрасной тревоги при беременности при попадании в группу риска по хромосомным аномалиям, нет риска спонтанного прерывания беременности. НИПТ одобрен во многих странах и включен в систему добровольного медицинского страхования (такие развитые страны как Швейцария, США, Канада, Нидерланды, Германия).
Какие же основные синдромы выявляет НИПТ
- Синдром Дауна (трисомия 21)
Частота встречаемости — 1:600 родов. Причем, риск трисомии 21 у плода увеличивается с возрастом женщины.
Одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями.
Существует ещё две формы данного синдрома:транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Y-хромосому) — 4% случаев, и мозаичный вариант синдрома — 5%.Характерные особенности, сопутствующие синдрому Дауна: «плоское лицо», аномальное укорочение черепа, кожная складка на шее у новорожденных, короткие конечности, открытый рот и другие признаки, которые может точно диагностировать врач-генетик.
- Синдром Эдвардса (трисомия 18)
Хромосомное заболевание характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом. Популяционная частота по миру 1:5000 на 2016 г.
Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии 21 хромосомы.
Основные признаки синдрома Эдвардса: малый вес, аномалии форм черепа, пороки сердца, дефекты межжелудочковой перегородки, умственная отсталость.
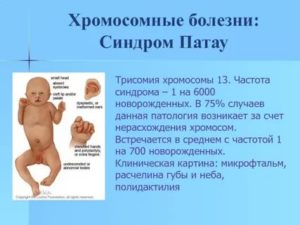
- Синдром Патау (трисомия 13)
Частота – 1:16000 родов. Характерны нарушения развития нервной системы, глаз, выраженная деформация лица, пороки развития сердца и других внутренних органов. Характерна глубокая задержка умственного развития. 90% детей умирают в возрасте до 1 года.
Гоносомные анеуплоидии:
- Синдром Шерешевского-Тернера
Если в клетке присутствует только одна хромосома вместо двух, это называется моносомией. Частота встречаемости – 1:2500.
Возникает в случае отсутствия второй половой хромосомы, встречается только у девочек. Единственная моносомия, при которой возможно рождение живого ребенка.
Хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом.
Частота – 1:500. Когда в клетках с мужским кариотипом присутствует дополнительная Х-хромосома. Встречается только у мальчиков. Это заболевание, при котором особи мужского пола имеют дополнительную Х-хромосому.
Обычно женщины имеют пару ХХ хромосом, а мужчины пару ХY хромосом, однако при этом заболевании мужчины имеют по крайней мере две Х-хромосомы и хотя бы одну Y хромосому.
Синдром Клайнфельтера является не только самой частой формой мужского гипогонадизма, бесплодия, эректильной дисфункции, но и одной из наиболее распространённых эндокринных патологий, занимая третье место после сахарного диабета и заболеваний щитовидной железы.
- Синдром XYY (синдром Джейкобса)
Частота – 1:1000. В клетках присутствует дополнительная Y-хромосома. Встречается только у мальчиков. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Наличие второй Y-хромосомы в большинстве случаев не ведёт к каким-либо физическим отклонениям.
В то же время, многие мужчины с XYY-синдромом имеют одну или несколько особенностей. При рождении они имеют нормальный рост, но часто быстрее растут в детстве. В среднем, во взрослом состоянии носитель выше, чем 75% мужчин того же возраста. Некоторые мужчины с синдромом XYY имеют небольшие нарушения координации движений, в результате чего могут казаться неуклюжими.
Фертильность чаще всего не нарушена, обычно такие мужчины гетеросексуальны и имеют нормальную сексуальную функцию. Тем не менее, описаны случаи существенного снижения фертильности, вплоть до бесплодия. У небольшого числа носителей также повышен уровень половых гормонов, связанных со сперматогенезом, что может вести к бесплодию ввиду нарушения образования спермы.
Неизвестно, насколько высоко число случаев бесплодия у мужчин с XYY-синдромом. IQ находится в пределах нормы, но часто несколько ниже, чем у родных братьев и сестёр. Примерно половина носителей имеет проблемы с обучением, в частности, могут быть нарушения и чтения.
Может быть повышен риск поведенческих проблем, таких как синдром гиперактивности, мужчины с XYY-синдромом часто импульсивны и эмоционально незрелы.
- Синдром тройной Х-хромосомы
Частота – 1:1000. В клетках присутствует 3 копии Х-хромосомы. Встречается только у девочек.
В большинстве случаев носители дополнительной X-хромосомы — женщины без заметных признаков патологии, поэтому при медицинских исследованиях 90% трисомиков по X-хромосоме остаются не выявленными.
Развитие может протекать с некоторыми нарушениями, могут возникнуть проблемы с координацией, моторикой и развитием речи. В некоторых случаях отмечен меньший размер головы (без заметного снижения умственных способностей).
Трисомия по X-хромосоме не приводит к значительным нарушениям фертильности, в большинстве случаев может проявляться только в более ранней менструации.Источник: https://medicalgenomics.ru/poleznoe/patologii-ploda-vo-vremya-beremennosti-sindromy.html
Трисомия — синдром лишней хромосомы: виды, причины, диагностика

Трисомией называют генетическую мутацию, при которой в кариотипе человека появляется дополнительная хромосома.
Хромосомы — это ядерные структуры, содержащие молекулу ДНК и предназначенные для хранения и передачи генетической информации. В соматических клетках человека каждая такая структура представлена двумя копиями.
Трисомия — это вид генетической патологии, при которой в клетках присутствуют три гомологичные хромосомы вместо двух. Такое нарушение происходит при оплодотворении и ведет к гибели плода либо к развитию тяжелых наследственных синдромов.
Поскольку на сегодняшний день не существует эффективных методов излечения таких заболеваний, крайне важная роль отводится пренатальной диагностике.
Из 23 хромосомных пар 22 идентичны у обоих полов, они называются аутосомами. 23-я пара представлена половыми хромосомами и различается у мужчин (XY) и у женщин (ХХ). Среди аутосомных нарушений чаще всего встречается трисомия по 21, 13 и 18-й хромосомам. Остальные патологии нежизнеспособны и приводят к самопроизвольному аборту на ранних сроках беременности.
На самом деле с фактом трисомии современный человек сталкивается достаточно часто, сам того не подозревая.
Какими бывают трисомии?
- Синдром трисомии 21-й хромосомы. Трисомию 21-й хромосомы называют синдромом Дауна. Он проявляется совокупностью различных патологий, основными из которых является нарушение интеллектуального развития, пороки сердечно-сосудистой и пищеварительной систем, а также специфический внешний вид.
Возможности современной медицины и педагогики позволяют таким людям интегрироваться в общество и вести активный образ жизни. При этом средняя продолжительность жизни у них составляет около 60 лет.
- Трисомия 18-й хромосомы. Синдром трисомии по 18-й хромосоме называется синдромом Эдвардса.
Это тяжелая патология, в большинстве случаев приводящая к преждевременным родам или самопроизвольным абортам. Даже если ребенок родился в срок, продолжительность жизни редко превышает один год.
- Клинически проявляется пороками развития центральной нервной системы, скелета и внутренних органов.
У таких детей диагностируется тяжелая умственная отсталость, микроцефалия, заячья губа, волчья пасть и множество других нарушений.
- Синдром Патау. Синдром Патау обусловлен трисомией 13-й хромосомы.
Клинически проявляется микроцефалией, нарушением развития ЦНС, тяжелой умственной отсталостью, пороками сердца, транспозицией сосудов, множественными пороками внутренних органов. Продолжительность жизни зависит от формы синдрома. В среднем она не превышает одного года, хотя 2–3% таких детей доживают до десяти лет.
- Трисомии половых хромосом.
Синдромы трисомии половых хромосом имеют более мягкое проявление, без угрозы жизни и инвалидизирующих пороков развития. Как правило, у таких пациентов нарушена репродуктивная функция, и может диагностироваться интеллектуальная недостаточность разной степени. В связи с этим они могут иметь проблемы с поведением и социализацией.
Все приведенные тримосии являются аутосомными, а все другие варианты – нежизнеспособные. Даже если в процессе развития происходит трисомия к какой-то другой хромосоме, то плод погибает еще внутриутробно, обычно на раннем сроке и может выглядеть как обычный выкидыш. Жизнеспособными являются только те зародыши, у которых трисомия произошла к хромосиме X или Y, причем в этом случае любые улинические проявления трисомии могут быть очень слабо выраженными.
Причины трисомии
Чаще всего трисомия возникает вследствие нарушения процесса расхождения гомологичных хромосом еще в анафазе первого мейоза. Результатом этого нарушения становится то, что в одну и ту же дочернюю клетку попадают сразу две гомологичные хромосомы, а вот во вторую дочернюю клетку – ни одной, то есть клетка становится нулисомной.
В некоторых случаях бывает так, что трисомия проявляется из-за патологии расхождения хроматид уже во втором мейозе. Это проявляется таким образом, что в одну гамету попадают сразу две идентичные хромосомы. Если оплодотворение произойдет при участии нормального спермия, то получится трисомная зигота. В этом случае данную патологию называют нерасхожденными хромосомами.
В большинстве случаев аутосомные трисомии становятся следствием нерасхождения хромосом, произошедшего еще в оогенезе, хотя теоретически это может произойти и в сперматогенезе. Нерасхождение может случиться и на ранних стадиях дробления уже оплодотворенной яйцеклетки, но это бывает значительно реже.
Другой причиной трисомии является мутация, возникшая уже после оплодотворения, на ранних этапах эмбриогенеза. В этом случае только часть клеток будет аномальный набор хромосом. Такое состояние называется мозаицизмом и протекает более благоприятно, чем синдром полной трисомии. Диагностировать данную патологию трудно, особенно в рамках пренатальной диагностики.
Развитие трисомий носит случайный характер и слабо связано с факторами окружающей среды, состоянием здоровья человека.
Точно сказать, почему происходит нерасхождение хромосом пока нельзя. Несмотря на то, что считается, что с возрастом риск родить ребенка с синдромом Дауна увеличивается, точно говорить о 100 % закономерности пока нельзя.
Безусловно, беременности в возрасте после 30 лет может быть опасной, и частота случаев рождения детей-даунов выше, чем у рожениц до 30 лет.Именно поэтому во время беременности рекомендуют проводить специальные анализы для выявления синдрома Дауна, ведь этот диагноз может быть показание к прерыванию беременности даже на поздних сроках.
Синдром делеции 22 хромосомы (синдром ДиДжорджи) у детей. Клинические рекомендации
- Аутоиммунные осложнения
- Велофарингеальная недостаточность
- Врожденный порок сердца
- Гипокальциемия
- Гипопаратиреоз
- Делеция 22 хромосомы
- Дефект Т-клеточного звена
- Задержка речевого и психомоторного развития
- Задержка физического развития
- Внутривенные иммуноглобулины
- Инфекционные осложнения
- Расщепление неба и верхней губы
- Синдром ДиДжорджи
АИГА – аутоиммунная гемолитическая анемия
АЛТ — аланинаминотрансфераза
АСТ — аспартатаминотрансфераза
ВВИГ — внутривенные иммуноглобулины
ВЗК – воспалительные заболевания кишечника
ВПС – врожденный порок сердца
ГКС — глюкокортикостероиды
ДНК — дезоксирибонуклеиновая кислота
ЖКТ — желудочно-кишечный тракт
ИТП – иммунная тромбоцитопения
КМ — костный мозг
КТ — компьютерная томография
ЛОР — ларингооторинолог
ЛПУ — лечебно-профилактическое учреждение
МЗ — Министерство здравоохранения
МКБ-10 — Международная классификация болезней 10-го пересмотра
МРТ —магнитно-резонансная томография
РКИ — рандомизированные контролируемые исследования
РНК — рибонуклеиновая кислота
РФ — Российская Федерация
Синдром del 22q11 – синдром делеции 22 хромосомы=синдром ДиДжорджи
СДД — синдром ДиДжорджи
ТГСК – трансплантация гемопоэтических стволовых клеток
УЗИ – ультразвуковое исследование
ЭКГ – электрокардиограмма
ЭхоКГ -эхокардиография
ЮРА – ювенильный ревматоидный артрит
del 22q11.2 – делеция длинного плеча 22 хромосомы локус 11.2
САТСН 22 — Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia, 22q deletion – порок сердца, лицевые аномалии, гипоплазия тимуса, расщелина неба, гипокальцемия, делеция 22
FISH –fluorescent in situ hybridization — флуоресцентная гибридизация in situ
ТВХ 1 ген –Т бокс 1 ген
TREC — T-cell Receptor Excision Circles
Термины и определения
Внутривенные иммуноглобулины – препараты, содержащие преимущественно нормальный человеческий IgG. Изготовляются из пулированной плазмы тысяч здоровых доноров, с применением специальных методов очистки и вирусинактивации.
Делеция – потеря участка хромосомы
Хромосомные аберрации – изменение числа и\или структуры хромосом
Микрогнатия — недоразвитие (гипоплазия) челюстных костей.
Ретрогнатия — смещение челюстной кости в дорзальном направлении (кзади)
Гипертелоризм — увеличенное расстояние между глазами
TREC – кольцевые фрагменты ДНК, образующиеся при развитии Т лимфоцитов в тимусе, в частности, в процессе формирования Т клеточного рецептора. Их концентрация в крови отражает эффективность тимопоэза. Используется для скрининга Т клеточных иммунодефицитов.
Трансплантация гематопоэтических стволовых клеток – метод лечения некоторых наследственных и приобретенных гематологических, онкологических и иммунных заболеваний, основанный на замене собственного, патологического кроветворения больного на нормальное кроветворение донора.
1.1 Определение
Синдром делеции 22-й хромосомы (синдром del 22q11) или синдром ДиДжоржи (СДД) — это совокупность морфологических, иммунологических и неврологических изменений, которые являются следствием делеции длинного плеча одной копии 22-й хромосомы — del 22q11.2 [1,2].
В классическом понятии этот синдром представляет собой комплекс симптомов, состоящий из патологии лицевого скелета (расщелины твердого неба), врожденного порока сердца, иммунодефицита вследствие гипоплазии (аплазии) тимуса и гипокальциемии, как результат гипоплазии паращитовидной железы [1,3].
Как ни один другой синдром, синдром del 22q11.2 вариабелен в количестве признаков и степени их выраженности, что и объясняет тот факт, что этот синдром в литературе имеет порядка десятка различных названий, включая синдром ДиДжорджи, САТСН 22, велокардиофациальный сидром, Шпринтцена синдром, Кайлера синдром, синдром лицевых и конотрункальных аномалий и т.д.[1,3,4].
1.2 Этиология и патогенез
В основе заболевания лежит нарушение формирования органов, происходящих их третьей жаберной дуги (нижняя часть лицевого скелета, тимус, паращитовидная железа, верхние отделы сердца и магистральных сосудов). Цитогенетические и молекулярные исследования показали, что делеция 22q11.
2 является ведущей причиной СДД и возникает спорадически более чем в 90% случаев [5,6,7]. В 10% случаев делеция наследуется от одного из родителей, так как наследование происходит аутосомно- доминантным путем [1,4].
В редких случаях синдром является проявлением перестроек других хромосом, а также мутации гена ТВХ1 [4].
Анализ ДНК пациентов с СДД хромосомы выявил, что в 85-90% случаев выпадающий участок является одним и тем же. Дефект находится между D22S427 на 22q11.21 и D22S801 на 22q11.23. В этом участке локализовано не менее 40 генов, что составляет около 3 млн пар нуклеиновых оснований.
В 10-12% случаев встречаются более короткие делеции, которые составляют 1,5-2 млн парных оснований. Было описано несколько пациентов с синдромом делеции 22-й хромосомы, имеющих делеции за пределами наиболее часто выпадающих участков [5,6].
Результаты проведенных исследований свидетельствуют о том, что степень выраженности фенотипа не коррелирует с размером делеции, т.е. пациент с потерей 1,5 млн парных оснований может иметь такой же по тяжести фенотип, как и с делецией в 3 млн парных оснований [2,5].
Кроме того, было замечено, что вариабельность фенотипических проявлений варьирует как внутри одной семьи, так и между семьями, несмотря на идентичные участки делеции [5].Делеция вызывает выпадение участка, включающего ген ТВХ, ген фактора транскрипции, участвующего в развитии фарингеальных дуг [5,6]. Эти изменения, в свою очередь, ведут к нарушению формирования сердца и магистральных сосудов, иммунологическим изменениям, расщеплению нёба и верхней губы, гипопаратиреоидизму, задержке умственного развития.
Несмотря на то, что ТВХ1, без сомнения, является главным геном, формирующим фенотип при синдроме делеции 22-й хромосомы, в результате исследований были выявлены и другие гены, недостаточная экспрессия которых может играть роль в формировании фенотипических проявлений [6,7].
Учитывая результаты работ по выяснению молекулярных основ заболевания, ясно, что в формировании фенотипа играет роль комплексное нарушение экспрессии и взаимодействия генов, их модификаторов и других составляющих, что приводит к дальнейшему нарушению эмбрио- и органогенеза [4,5,7].
Соответственно, при отсутствии или нарушении функции и экспрессии генов и дальнейших процессов происходит формирование пороков развития, характерных для СДД [1,5].
1.3 Эпидемиология
Синдром делеции 22-й хромосомы — одна из самых частых делеций среди других хромосомных аберраций в человеческом геноме, по частоте она уступает лишь синдрому Дауна, трисомии по 21-й хромосоме. Частота встречаемости варьирует от 1:4000 до 1:6000 новорожденных [1,2,3].
Не наблюдается ни половой, ни этнической предрасположенности к данному синдрому. Большинство пациентов с СДД имеют патологию лицевого скелета и врожденный порок сердца и развивают гипокальциемию вскоре после рождения [6].
Пациенты, не имеющие данных симптомов, зачастую диагностируются в раннем возрасте, и правильный значительно запаздывает.
1.4 Кодирование по МКБ-10
Другие иммунодефициты (D84):
D84.1 – Синдром ДиГеорга.
1.5 Классификация
Исторически сложилось, что в литературе часто используется разделение синдрома на полный и неполный (частичный) [1,3,5]:
- Термин «Полный синдром ДиДжорджи» использовался у пациентов, имеющих полный спектр типичных проявлений, включая выраженный иммунодефицит.
- Термин «Частичный синдром ДиДжорджи» использовался у пациентов, если они имели лишь некоторые типичные признаки, особенно без проявлений выраженного иммунодефицита. Частичный синдром делеции 22-й хромосомы в значительной степени превалирует по количеству в сравнении с полным.
2.1 Жалобы и анамнез
Спектр клинических проявлений при синдроме делеции 22-й хромосомы достаточно широк [3,6,9,11,12,14,15], поэтому жалобы и анамнез заболевания могут быть крайне разнообразными и различными по степени выраженности:
- Врожденный порок сердца представлен не менее, чем в 80% случаев. Некоторые из пороков являются более патогномоничными: прерывание дуги аорты, общий артериальный ствол и тетрада Фалло являются наиболее частыми среди данной группы детей [6,11].
- Гипокальциемия/гипопаратиреоз может проявляться судорожным синдромом при выраженном дефиците кальция в младенческом возрасте [6,12].
- Поражение носоглоточного аппарата выявлено примерно в 70% случаев и проявляется в виде велофарингеальной недостаточности, расщеплении нёба, губы, раздвоении уздечки нёба, гнусавым оттенком голоса, также описано снижение обоняния, кондуктивная и/или сен- соневральная тугоухость [6,10,13].
- Характерные черты лица (удлиненное лицо, микрогнатия или ретрогнатия, широкая переносица, мелкие зубы, асимметрия лица при плаче, опущенные вниз уголки рта, глазной гипертелоризм, низко посаженные и деформированные ушные раковины, бульбообразный кончик носа) [2,5,10].
- Иммунологические нарушения встречаются в 77% случаев. Однако инфекционные проявления вследствие дефекта иммунной системы дебютируют не с рождения. Чаще других звеньев поражается Т-клеточное звено, что проявляется предрасположенностью к грибковым заболеваниям, пневмоцистной инфекции, некоторым бактериальным и вирусным инфекциям [1,8,10].
- Нарушение выработки Т-клеток может предрасполагать к аутоиммунным заболеваниям. Описаны такие осложнения синдрома делеции 22-й хромосомы, как ЮРА, ХТП, АИГА, ВЗК, болезнь Грейвса, аутоиммунный увеит, бронхиальная астма [1,9,10,14].
- Задержка физического развития нередко наблюдается у пациентов с синдромом делеции 22q11.2-й хромосомы, которые несколько отличаются от стандартных таблиц [1,2,6,10].
- Задержка речевого и психомоторного развития наблюдается у 70—90% и проявляется с возрастом, однако тестирование пациентов с задержкой развития имеет смысл только в случаях сочетании с другими признаками [2,10,15].
2.2 Физикальное обследование
Физическое развитие большинства пациентов низкое и дисгармоничное по весу [1,10].
Стигмы дисэмбриогенеза широко вариабельны и не являются патогномоничными, однако чаще других признаков обращают на себя внимание глазной гипертелоризм, бульбообразный кончик носа и низко посаженные и\ или деформированные ушные раковины [6,10,13].
Могут проявляться признаки дыхательной и сердечной недостаточности [2,11]. Могут встречаться пороки развития дыхательной, пищеварительной, костно-мышечной и других систем.
Задержка умственного и речевого развития встречается у подавляющего числа пациентов с данным синдромом [2,3,15].
2.3 Лабораторная диагностика
- Рекомендуется клинический анализ крови [2,3].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 3)
- Рекомендуется исследование уровня ионизированного кальция, паратиреоиного гормона [12].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2)
- Рекомендуется исследование уровня гормонов щитовидной железы [12].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3)
- Рекомендуется исследование уровня сывороточных иммуноглобулинов и клеточного иммунитета, включая определение количества наивных Т-лимфоцитов [1,10,16,17].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2)
Источник: https://medi.ru/klinicheskie-rekomendatsii/sindrom-deletsii-22-khromosomy-sindrom-didzhordzhi-u-detej_14268/
Основные виды трисомии и причины ее возникновения
Трисомия — это числовые нарушения хромосом в результате которых в гомологичной паре присутствует одна лишняя хромосома. В норе у человека 46 хромосом, которые распределены по парам, всего 23 пары. Помимо трисомии могут возникать процессы, при которых наоборот недостает одной хромосомы из пары, моносомии.
Основные причины возникновения хромосомной мутации
Причины трисомии активно изучаются. В связи с тем, что количество детей, рожденных с анеуплоидией, увеличивается, необходимо до конца понять механизм образования хромосомных патологий на ранних стадиях беременности.
Одна из причин проявляется в анафазе I мейоза, когда одна или несколько паргомологичных хромосом не разойдутся. Далее они направляются к одному и тому же центру клетки. В результате возникает гамета с числовыми нарушениями. Такое явление называется нерасхождение.
Чаще всего числовые мутации связаны с нарушениями в кариотипе яйцеклетки. Если рассматривать риски возникновения аномалий в сперотозоиде и яйцеклетки, то на долю яйцеклеток приходится 75%, против 25% у сперматозоидов. Считается, что после 35 лет у женщины риск родить ребенка с анеуплоидией составляет 1%.
Генетические хромосомные заболевания могут иметь наследственных характер, если кто- то из родителей или родственников является носителем патологий. Сейчас учащаются случаи рождения детей с хромосомными у здоровых родителей. К основными причинам, увеличивающим вероятность возникновения болезни относятся:
- Возраст женщины более 35 лет. Это связано со снижением качества яйцеклеток.
- Возраст мужчины более 40 лет. Считается, что после этого рубежа качество сперматозоидов заметно снижается.
- Наличие патологий у родителей или родственников.
- Наличие прерывания беременности, выкидыша, замершей беременности.
- Перенесение тяжелых инфекционных заболеваний в период зачатия и на первых стадиях беременности.
Диагностирование аномалий должно протекать в несколько этапов:
- Визуальная диагностика с помощью УЗИ. Если врач наблюдает, например, отсутствие носовой перегородки или лишние шейные складки, то необходимо делать биохимию.
- Биохимический скрининг. Выявляется превышение концентрации специфических белков в крови матери.
- Неинвазивный пренатальный тест (НИПТ). Является безопасным методом генетического анализа на все виды хромосомных мутаций по крови матери.
- Инвазивное тестирование. Обычно назначается после выявления рисков при НИПТ или биохимии. Заключается в биопсии материала плода, хориона или амниотической жидкости и дальнейшем ДНК тесте.
Прерывание беременности назначается только врачом и желательно на основании инвазивного анализа.
Какие разновидности бывают?
К основным видам аномалий можно отнести аутосомные и половые трисомии. К первому типу относятся три основных патологии:
- Синдром Дуана (трисомия 21 хромосомы). Наиболее часто встречающийся. По разным данной частота рождения детей с синдромом Дауна 1 на 1000-3000 малышей. Проявляется широко посаженными глазами, косоглазием, широкие лоб и переносица, измененными ушными раковинами, складки у глаз и на шее. Часто сопровождается слабоумием и нарушением работы внутренних органов.
- Синдром Патау (трисомия 13). Встречается реже, чем Даун примерно в 10 раз. Проявляется расщеплением неба и верхней губы, ушные раковины посажены низко и изменены, узкие глазные щели, деформация стоп, короткая шея с лишним складками. Сопровождается слабоумием. Системы и органы работают с нарушениями.
- Синдром Эдвардса (трисомия 18). Как и Патау встречается редко. К внешним признакам относится: изменение формы черепа, отсутствие ушных мочек, деформация ушных раковин, маленькие глаза, короткий большой палец, расщепление неба, стопа-качалка. В основном присутствуют пороки сердца и желудочно-кишечного тракта. Слабоумие.
- Помимо удвоения аутосомных хромосом встречаются и анеуплоидии половых (АПХ), в т.ч. и трисомии:
- Синдром Клайнфельтера. Заболевание, связанное с удвоение X хромосомы в паре мужских хромосом XY с образованием кариотипа XYY. Болеют только мужчины. Проявляется высоким ростом, длинными конечностями и худощавым телосложением. Вторичные половые признаки выражены слабо. Страдают слабоумием и шизофреническими припадками. Бесплодны.
- Синдром тройной X. Наблюдается у женского пола. Определяется утроением половой X хромосомы, образуют кариотип XXX. Встречается довольно часто 1 на 1200 малышей. Проявляется высоким ростом, слабоумием, задержкой речи, маленькие или кривые пальцы, заболевания мочеполовой системы.
- Синдром супермена или двойной Y. Встречается Только у мужчин с образованием лишней Y, образуя кариотип XYY. Имеют рост выше среднего, удлиненные конечности. Умственные способности занижены, задержка речи.
В отличие от трисомии 21, 13, 18, с АПХ люди вполне жизнеспособны и могут иметь потомство. Внешний вид не сильно выделяется.
Можно ли вылечить заболевание?
К сожалению, генетические числовые хромосомные нарушения не поддаются лечению. Их можно диагностировать на ранних стадиях беременности и вовремя принять меры. Для профилактики возникновения плода с патологией, рекомендуется пройти полное клиническое обследование перед зачатием. Если женщина и/или мужчина в группе риска (см. выше),то стоит уделить этому вопросу особое внимание.
В отличие от Дауна, Эдвардса и Патау, с АПХ можно вести полноценную жизнь с помощью гормональной терапией
Источник: https://InLab-Genetics.ru/articles/osnovnye-vidy-trisomii-i-prichiny-ee-vozniknovenija/